
Volume 53 - Issue 2 - April 2005
Contents
Research Article
Surface enthalpy of goethite
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 113-122
-
- Article
- Export citation
Adsorption of volatile organic compounds onto porous clay heterostructures based on spent organobentonites
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 123-136
-
- Article
- Export citation
Sorption of 3-amino-1,2,4-triazole and Zn(II) onto montmorillonite
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 137-146
-
- Article
- Export citation
Atomic force microscopy study of montmorillonite dissolution under highly alkaline conditions
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 147-154
-
- Article
- Export citation
Conversion of chrysotile to a magnesian smectite
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 155-161
-
- Article
- Export citation
-
Chrysotile from Thetford Mines in Quebec, Canada was treated first with mild formic or oxalic acid at concentrations of 0.5 to 2.0 N at 200°C in Teflon-lined 12.0 mL Parr bombs. The reaction products were identified by X-ray diffraction as a poorly crystalline Fe-bearing kerolite-like 2:1 layer silicate (which will be described as a kerolitic precipitate or a kerolitic mesophase in this report). Electron microscopic examination showed a thin foily morphology for this kerolitic mesophase that may have formed by the following reaction:(1)
$\mathop {{\rm{M}}{{\rm{g}}_{\rm{6}}}{\rm{S}}{{\rm{i}}_{\rm{4}}}{{\rm{O}}_{{\rm{10}}}}{{\left( {{\rm{OH}}} \right)}_{{8_{\left( {\rm{s}} \right)}}}}}\limits_{{\rm{chrysotile}}} + 6.02{{\rm{H}}^{\rm{ + }}}_{\left( {{\rm{aq}}} \right)} + 0.54{\rm{F}}{{\rm{e}}^{{\rm{2 + }}}}_{\left( {{\rm{aq}}} \right)} \to \mathop {\left( {{\rm{M}}{{\rm{g}}_{{\rm{2}}{\rm{.46}}}}{\rm{Fe}}_{{\rm{0}}{\rm{.54}}}^{{\rm{2 + }}}} \right){\rm{S}}{{\rm{i}}_{\rm{4}}}{{\rm{O}}_{{\rm{10}}}}{{\left( {{\rm{OH}}} \right)}_{\rm{2}}} \cdot n}\limits_{{\rm{kerolitic}}\;{\rm{mesophase}}} {{\rm{H}}_{\rm{2}}}{{\rm{O}}_{\left( {\rm{s}} \right)}} + {\rm{3}}{\rm{.55M}}{{\rm{g}}^{{\rm{2 + }}}}_{\left( {{\rm{aq}}} \right)}$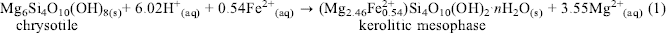
The magnetite impurity in the initial chrysotile asbestos served as the source of Fe in the above reactions. Subsequently, this kerolitic precipitate was reacted with 0.2 N NaOH for 48–96 h at 200°C and a highly crystalline smectite was formed with the same foily morphology as the kerolitic precipitate. X-ray spectral analyses of the kerolitic mesophase and smectite suggest the following reaction to have taken place:(2)
$\eqalign{ & \mathop {\left( {{\rm{M}}{{\rm{g}}_{{\rm{2}}{\rm{.46}}}}{\rm{Fe}}_{{\rm{0}}{\rm{.54}}}^{{\rm{2 + }}}} \right){\rm{S}}{{\rm{i}}_{\rm{4}}}{{\rm{O}}_{{\rm{10}}}}{{\left( {{\rm{OH}}} \right)}_{\rm{2}}}}\limits_{{\rm{kerolitic}}\;{\rm{mesophase}}} \cdot n{{\rm{H}}_{\rm{2}}}{{\rm{O}}_{\left( {\rm{s}} \right)}} + {\rm{0}}{\rm{.53M}}{{\rm{g}}^{{\rm{2 + }}}}_{\left( {{\rm{aq}}} \right)} + 0.54{\rm{NaO}}{{\rm{H}}_{\left( {{\rm{aq}}} \right)}} + 0.01{\rm{F}}{{\rm{e}}^{{\rm{3 + }}}}_{\left( {{\rm{aq}}} \right)} \to \cr & \;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\mathop {\;\;{\rm{N}}{{\rm{a}}_{0.54}}\left( {{\rm{M}}{{\rm{g}}_{{\rm{2}}{\rm{.99}}}}{\rm{Fe}}_{{\rm{0}}{\rm{.01}}}^{{\rm{2 + }}}} \right)\left( {{\rm{S}}{{\rm{i}}_{{\rm{3}}{\rm{.46}}}}{\rm{Fe}}_{{\rm{0}}{\rm{.54}}}^{{\rm{3 + }}}} \right){{\rm{O}}_{10}}{{\left( {{\rm{OH}}} \right)}_{{\rm{2}}\;\left( {\rm{s}} \right)}}}\limits_{{\rm{saponite}}} + 0.54{\rm{S}}{{\rm{i}}^{{\rm{4 + }}}}_{\left( {{\rm{aq}}} \right)} \cr} $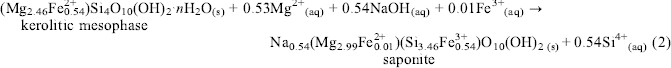
The reaction products, a kerolitic mesophase and smectite, possess a non-fibrous habit in contrast to the fibrous (asbestiform) morphology of chrysotile.
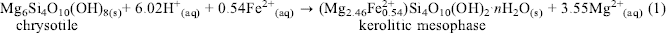
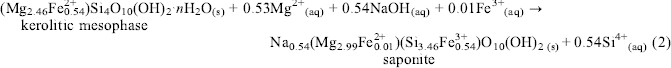
Characteristics of the minerals associated with gold in the Shewushan supergene gold deposit, China
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 162-170
-
- Article
- Export citation
Modeling the response of pyrophyllite interlayer to applied stress using steered molecular dynamics
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 171-178
-
- Article
- Export citation
Cation-site partitioning in Ti-rich micas from Black Hill (Australia): A multi-technical approach
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 179-189
-
- Article
- Export citation
The 2M1 dioctahedral mica polytype: A crystal chemical study
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 190-197
-
- Article
- Export citation
Announcement
Forthcoming papers
-
- Published online by Cambridge University Press:
- 01 January 2024, p. 198
-
- Article
-
- You have access
- Export citation