
Volume 54 - October 2006
Contents
Research Article
Mineralogy and Geochemistry of the Host-Rock Alterations Associated with the Shea Creek Unconformity-Type Uranium Deposits (Athabasca Basin, Saskatchewan, Canada). Part 1. Spatial Variation of Illite Properties
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 275-294
-
- Article
- Export citation
A New Ni-Rich Stevensite From the Ophiolite Complex of Othrys, Central Greece
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 653-666
-
- Article
- Export citation
FTIR investigation of the evolution of the octahedral sheet of kaolinite-smectite with progressive kaolinization
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 1-11
-
- Article
- Export citation
Crystal-Size Dependence of Illite-Smectite Isotope Equilibration with Changing Fluids
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 531-540
-
- Article
- Export citation
Calorimetric Determination of the Enthalpies of Formation of Hydrotalcite-Like Solids and Their Use in the Geochemical Modeling of Metals in Natural Waters
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 409-417
-
- Article
- Export citation
Crystal-Chemical Factors Responsible for the Distribution of Octahedral Cations Over trans- and cis-Sites in Dioctahedral 2:1 Layer Silicates
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 131-152
-
- Article
- Export citation
Nickel Solubility and Precipitation in Soils: A Thermodynamic Study
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 153-164
-
- Article
- Export citation
Controlled Polymerization of Metanilic Anion within the Interlayer of NiAl Layered Double Hydroxide
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 418-425
-
- Article
- Export citation
-
The controlled chemical oxidative polymerization of metanilic anion $(m{\rm{ - N}}{{\rm{H}}_2}{{\rm{C}}_6}{{\rm{H}}_4}{\rm{SO}}_3^ - )$
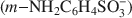 within the interlayer of NiAl layered double hydroxide was performed using, for the first time, ammonium persulfate as the oxidizing agent. The quantity of oxidizing agent required for control of the interlayer polymerization was investigated systematically and it was found that interleaved polyaniline sulfonic (PANIS) was present in different oxidation states and protonation levels when different quantities of external oxidizing agents were added. A mechanism for the oxidative polymerization of metanilic anion in NiAl layered double hydroxide is proposed, based on the intercalation of the oxidizing agent and the interlayer polymerization of monomer. The resulting PANIS/NiAl LDH composites were characterized by powder X-ray diffraction, ultraviolet-visible absorption spectra, Fourier transform infrared and X-ray photoelectron spectroscopy.
within the interlayer of NiAl layered double hydroxide was performed using, for the first time, ammonium persulfate as the oxidizing agent. The quantity of oxidizing agent required for control of the interlayer polymerization was investigated systematically and it was found that interleaved polyaniline sulfonic (PANIS) was present in different oxidation states and protonation levels when different quantities of external oxidizing agents were added. A mechanism for the oxidative polymerization of metanilic anion in NiAl layered double hydroxide is proposed, based on the intercalation of the oxidizing agent and the interlayer polymerization of monomer. The resulting PANIS/NiAl LDH composites were characterized by powder X-ray diffraction, ultraviolet-visible absorption spectra, Fourier transform infrared and X-ray photoelectron spectroscopy.
Mineralogy and Geochemistry of the Host-Rock Alterations Associated with the Shea Creek Unconformity-Type Uranium Deposits (Athabasca Basin, Saskatchewan, Canada). Part 2. Regional-Scale Spatial Distribution of the Athabasca Group Sandstone Matrix Minerals
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 295-313
-
- Article
- Export citation
Properties of surface-modified colloidal particles
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 12-24
-
- Article
- Export citation
Hydrothermal Synthesis of Mg-Rich and Mg-Ni-Rich Kaolinite
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 667-677
-
- Article
- Export citation
Clay Minerals Formed During Propylitic Alteration of a Granite and Their Influence on Primary Porosity: A Multi-Scale Approach
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 541-554
-
- Article
- Export citation
Comparison of K-Ar Ages of Diagenetic Illite-Smectite to the Age of a Chemical Remanent Magnetization (CRM): An Example from the Isle of Skye, Scotland
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 314-323
-
- Article
- Export citation
Adhered Zeolite Preparation on and Within a Muscovite Mica by Hydrothermal Growth
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 678-688
-
- Article
- Export citation
-
Zeolites and other open framework materials provide a powerful tool for remediation and solidification of a range of cationic wastes (e.g.${\rm{NH}}_4^ + $
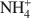 , Pb2+) due to the combined properties of large surface area and cation exchange capacity. However, practical barriers exist to the continued expansion of their use, including handling issues related to the fine particle size, and continued ion exchange following waste adsorption. This study examines the synthesis and characterization of zeolites adhered to a muscovite mica wafer, in order to assess if practical benefit can be derived from the preparation of layered composite materials. The paper demonstrates that increased metal adsorption, as demonstrated by surface chemical composition, can be induced in regions by growth of zeolite on and within the lamellar structure of the matrix. X-ray diffraction studies suggest that a site-specific crystallization mechanism controls the zeolite type and extent of growth, thereby reducing control over the zeolites prepared. However, although increased adsorption has been introduced to the mica, the amount of zeolite added is small (<50 mg per gram of muscovite), and thus any adsorption is very limited.
, Pb2+) due to the combined properties of large surface area and cation exchange capacity. However, practical barriers exist to the continued expansion of their use, including handling issues related to the fine particle size, and continued ion exchange following waste adsorption. This study examines the synthesis and characterization of zeolites adhered to a muscovite mica wafer, in order to assess if practical benefit can be derived from the preparation of layered composite materials. The paper demonstrates that increased metal adsorption, as demonstrated by surface chemical composition, can be induced in regions by growth of zeolite on and within the lamellar structure of the matrix. X-ray diffraction studies suggest that a site-specific crystallization mechanism controls the zeolite type and extent of growth, thereby reducing control over the zeolites prepared. However, although increased adsorption has been introduced to the mica, the amount of zeolite added is small (<50 mg per gram of muscovite), and thus any adsorption is very limited.
Sorption of Nitroaromatics by Ammonium- and Organic Ammonium-Exchanged Smectite: Shifts from Adsorption/Complexation to a Partition-Dominated Process
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 426-434
-
- Article
- Export citation
Geology, Mineralogy and Origin of Clay Minerals of the Pliocene Fluvial-Lacustrine Deposits in the Cappadocian Volcanic Province, Central Anatolia, Turkey
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 555-570
-
- Article
- Export citation
Use of atomic force microscopy for examining wet clay
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 25-28
-
- Article
- Export citation
Heating Fe Oxide-Rich Soils Increases the Dissolution Rate of Metals
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 165-175
-
- Article
- Export citation
The Smectite-to-Disordered Kaolinite Transition in a Tropical Soil Chronosequence, Pacific Coast, Costa Rica
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 571-586
-
- Article
- Export citation
Thermal and Mineral Properties of Al-, Cr-, Mn-, Ni- and Ti-Substituted Goethite
-
- Published online by Cambridge University Press:
- 01 January 2024, pp. 176-194
-
- Article
- Export citation